Наследственные полинейропатии
 Наследственные полинейропатии
Наследственные полинейропатии(информация для пациентов)
Что такое «Наследственные полинейропатии»?
Наследственные полинейропатии – это большая группа клинически разнообразных полинейропатий с возможным вовлечением различных органов и систем, в том числе и центральной нервной системы, которые развиваются в результате мутаций (поломок) в генах человека.
Какие мифы существуют среди населения относительно наследственных заболеваний?
- "Наследственные заболевания развиваются с рождения или в раннем детстве"
Это не так - дебют наследственной болезни возможен и во взрослом возрасте, в том числе и после 50 лет. Более того, зачастую пациенты не могут точно назвать возраст начала заболевания, так как симптомы развиваются незаметно. - "Наследственная патология должна быть и у близких родственников. А если в роду ни у кого нет, значит и у меня тоже нет".
Это не так - наследственные заболевания передаются разными путями: по аутосомно-доминантному, аутосомно-рецессивному, Х-сцепленному типу, также возможен митохондриальный тип наследования и др. Часто в семье есть асимптомные носители мутантного гена без клинических признаков заболевания, а симптомы болезни могут быть только у одного из членов семьи. Поэтому отсутствие отягощенного семейного анамнеза не исключает наследственный генез заболевания.
 Какие бывают «Наследственная полинейропатии»?
Какие бывают «Наследственная полинейропатии»?Наследственных полинейропатий очень много, причиной из развития могут быть мутации более чем в 100 генах. Классифицируют наследственные нейропатии в основном по типу наследования, вовлечению тех или иных нервных волокон и характеру их поражения.
Ниже представлена лишь часть из них, преимущественно тех, которые на практике могут диагностироваться в возрасте старше 20 лет:
- Наследственные моторно-сенсорные нейропатии (НМСН)
- Наследственная нейропатия со склонностью к параличам от сдавления (ННСПС)
- Наследственные сенсорные и автономные нейропатии (НСАН)
- Болезнь Фабри
- Транстиретиновая семейная амилоидная нейропатия (ТТР-САП)
 Как часто диагностируются наследственные полинейропатии среди населения?
Как часто диагностируются наследственные полинейропатии среди населения?Частота встречаемости всех форм НМСН варьирует от 10 до 40 случаев на 100 000 населения в различных популяциях. На НМСН 1 типа приходится до 70% всех случаев НМСН. Распространенность ННСПС 2-5 случая на 100 000 населения. Болезнь Фабри - 1 случай на 40 000 - 60 000 мужчин. По примерным подсчетам в США встречаемость ТТР-САП составляет 1 случай на 100 000 человек. Следует отметить, что настороженность относительно дебюта наследственной патологии во взрослом возрасте среди врачей разных специальностей достаточно низкая, поэтому некоторые цифры распространенности могут быть занижены.
Какие симптомы наблюдаются у пациентов с наследственными полинейропатиями?
Наследственные заболевания периферических нервов очень разнообразны по клиническим проявлениям. Ниже представлены данные некоторых из них.
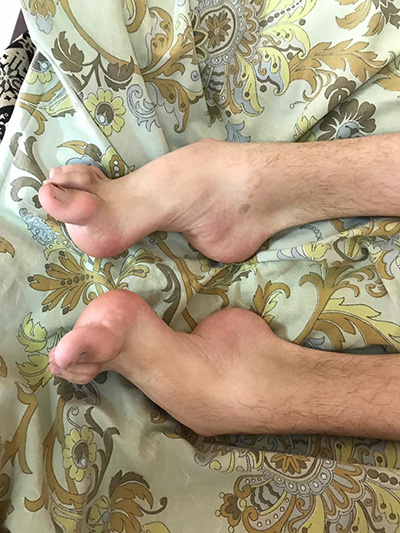
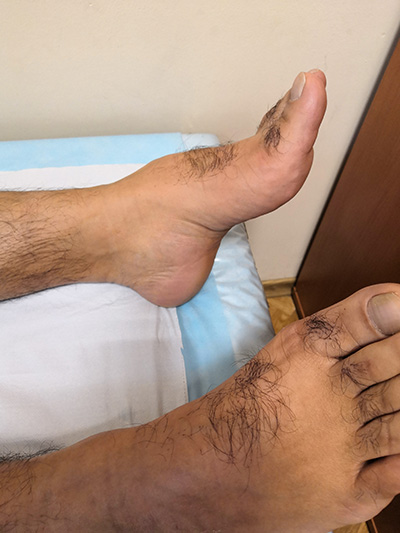
Наследственная моторно-сенсорная нейропатия (НМСН) I типа (болезнь Шарко-Мари-Тута) – наиболее часто диагностируемая (в том числе во взрослом возрасте) наследственная полинейропатия, в основе развития которой лежит дефект гена PMP22, в результате чего повреждается миелиновая оболочка периферических нервов. Заболевание чаще всего имеет аутосомно-доминантный тип наследования. Соотношение мужчин и женщин с НМСН 1 типа примерно равно. Симптомы заболевания появляются на первом десятилетии жизни в 75% случаев, в начале второго десятилетия реже - до 10-25%. Обычно чем позднее манифестирует заболевание, тем более благоприятно оно протекает.
Клиническая картина НМСН 1 типа:
- пациентов беспокоят болезненные спазмы мышц голеней (крампи), слабость и деформация стоп, изменение походки, затруднения при беге или подъеме по лестнице;
- постепенно слабость развивается в кистях, в результате чего появляются затруднения при застегивании пуговиц, открывании двери ключом и т.д. В целом руки вовлекаются не ранее чем через 10 лет после появления первых симптомов болезни;
- в меньшей степени беспокоит онемение кистей и стоп, часто эти изменения игнорируются пациентом.
При осмотре обращает внимание наличие деформации стоп, контрактур ахилловых сухожилий.
Клиническая картина других типов НМСН и НСАН вариабельна и определяется генетическим дефектом.
Наследственная нейропатия со склонностью к параличам от сдавления (ННСПС) - также часто диагностируемая во взрослом возрасте наследственная нейропатия (дебютирует, как правило, на 2–3-м десятилетии жизни), обусловленная мутацией в гене PMP22, характеризуется повышенной "чувствительностью" периферических нервов к сдавлению в костно-фиброзных каналах (туннелях), что приводит к повторяющимся эпизодам компрессионных туннельных мононевропатий.
Возможно поражение любого периферического нерва, но чаще всего сдавлению подвергаются:
- лучевой нерв на уровне плеча (спиральный канал), при этом развивается слабость разгибателей кисти, кисть "висит" как плеть, возникает онемение наружного края предплечья и кисти;
- малоберцовый нерв на уровне коленного сустава (фибулярный канал), при этом развивается слабость разгибателей стопы, стопа начинает "шлепать", возникает онемение наружного края голени и стопы;
- локтевой нерв на уровне локтевого сустава (кубитальный канал), при этом развивается слабость межкостных мышц кисти, мышцы - отводящих мизинец и других, возникает онемение 4 и 5 пальцев кисти;
- срединный нерв на уровне запястья (карпальный канал), при этом развивается слабость мышц возвышения большого пальца, возникает онемение 1-3 пальцев кисти, характерен болевой синдром;
- и т.д. с развитием соответствующей клинической картины поражения того или иного нерва.
Развитию симптомов, как правило, предшествует незначительная травма, нахождение длительное время в неудобной статической поза (на четвереньках, на корточках, облокотившись на локоть и др.), ношение неудобной одежды, непривычная физическая нагрузка и прочее.
Мышечная слабость, как правило, сохраняется в течение нескольких дней или недель, а затем сила мышц постепенно восстанавливается (в течение нескольких недель или месяцев). Эпизоды "защемлений" нервов имеют тенденцию к повторению (всего у пациентов может быть от 1 до 10 таких эпизодов). Следует учитывать, что часто лечащие врачи не задумываются о том, что причиной рецидивирующих (повторяющихся) туннельных невропатий – является наследственная патология, что приводит к поздней диагностике и неоправданным медицинским вмешательствам (операциям по поводу туннельных невропатий).
Болезнь Фабри - редкое генетическое заболевание, обусловленное мутацией гена GLA, картированного на длинном плече хромосомы Хq 22.1 и контролирующего структуру фермента альфа-галактозидазы А. Нередко начало заболевания отмечается в подростковом возрасте. Тип наследования - сцепленное с Х-хромосомой.
Клиническая картина:
- жгучая колющая боль в ладонях и стопах; острые приступы мучительной боли в кистях и стопах (кризы Фабри);
- нарушение потоотделения; непереносимость жары/холода;
- сыпь на коже (ангиокератомы);
- помутнение роговицы в виде завитка, которое не ослабляет зрение;
- шум в ушах, потеря слуха;
- желудочно-кишечные расстройства, диарея;
- кардиологические проявления (включая увеличенное сердце и нарушение ритма);
- нарушение функции почек, которое в конечном итоге приводит к терминальной стадии хронической почечной недостаточности;
- нарушение мозгового кровообращения (чаще ишемический инсульт, но может быть и внутримозговое кровоизлияние, или венозный тромбоз).
Существуют “красные флаги” заболевания: отягощенный семейный анамнез, автономные нарушения (ортостатическая гипотензия, эректильная дисфункция, нарушение потоотделения), нарушение работы органов пищеварительного тракта (запоры, диарея, тошнота) и мочеиспускания, поражение сердца (рестриктивная кардиомиопатия, аритмии, блокады сердца), синдром карпального канала (идиопатический, часто двусторонний), потеря массы тела, помутнение стекловидного тела, прогрессирующая полинейропатия:
- при раннем дебюте заболевания (<40 лет) в первую очередь поражаются дистальные тонкие волокна болевой и температурной чувствительности, что клинически выражается наличием парестезий, аллодинии, гипо-/гипералгезии, боли, снижением температурной чувствительности;
- поражение толстых миелинизированных чувствительных и двигательных волокон присоединяется позднее, приводя к нарушению тактильной и проприоцептивной чувствительности, слабости в конечностях, мышечной атрофии.
На основании чего устанавливается диагноз "Наследственная полинейропатия"?
Иногда клиническая картина и симптомы настолько яркие, что заболевание можно поставить "от двери", и после консультации сразу назначить генетическое обследование. Однако на практике чаше всего приходится проводить диагностику в несколько этапов.
На первом этапе нужно понять: есть полинейропатия или нет. Для подтверждения генерализованного поражения периферических нервов необходимы:
- анализ истории развития заболевания
- оценка неврологического статуса
- электронейромиография (ЭНМГ)
Проведение ЭНМГ-исследования необходимо во всех случаях, поскольку данный метод позволяет не только подтвердить поражение периферических нервов, но и уточнить характер их повреждения, следовательно, определить тип нейропатии. Важно проведение исследования хорошо подготовленным и опытным специалистом на миорафе высокого класса. Методологические ошибки и недостаточный объем данного исследования часто приводят к ошибочным диагнозам. Поэтому мы рекомендуем проведение ЭНМГ в нашем центре.
На втором этапе утоняется причина полинейропатии, исключаются те, для которых разработана эффективная терапия. Для выявления наиболее часто диагностируемых во взрослом возрасте приобретенных полинейропатий назначаются следующие лабораторные и инструментальные обследования:
- общий клинический и развернутый биохимический анализы крови;
- RW, анти-ВИЧ, НВsAg и анти-HCV;
- общий анализ мочи;
- анализ крови на уровни витаминов группы В, гомоцистеин;
- электрофорез белков сыворотки и мочи с иммунофиксацией;
- УЗИ периферических нервов;
- анализ цереброспинальной жидкости (ликвор) и другие.
- уровень фитановой кислоты в крови методом хроматомасс-спектрометрии (повышение при болезни Рефсума);
- определение активности лизосомного фермента альфа-галактозидазы А, в том числе с помощью флюориметрического метода («сухое» пятно) - исключение болезни Фабри;
- биопсия икроножного нерва, малых слюнных желез и абдоминального жира – для выявления отложения амилоида, характерного для ТТР-САП).
- Однако окончательный диагноз той или иной наследственной полинейропатии может быть подтвержден только по результатам генетического анализа. Оптимальным является генетическое обследование по следующему алгоритму:
- при убедительной клинико-анамнестической и параклинической картине в пользу той или иной наследственной полинейропатии проводится прицельная генетическая диагностика одного гена, в том числе с использованием скрининг тестов методом "сухого пятна";
- если подозревается наследственный генез полинейропатии, тип её не определен целесообразно проведение более широкого генетического анализа - секвенирования нового поколения (NGS).
Какое существует лечение при наследственной полинейропатии?
Важно помнить, что для некоторых наследственных полинейропатий, ассоциированных с известным метаболическим дефектом, разработана терапия:
- при болезни Фабри - это фермент-заместительная терапия рекомбинантными препаратами альфа-галактозидазы А
- при ТТР-САП – стабилизация молекулы транстиретина (Тафамидис), трансплантация печени;
- при порфирийной полинейропатии - введение аргината гема;
- при болезни Рефсума – диетотерапия с ограничением поступления фитановой кислоты (исключение потребления зеленых овощей, говядины, рыбы (тунец, пикша, треска)), плазмаферез.
При некоторых наследственных нейропатиях, для которых характерно поражение органов и систем, важно мониторировать функцию сердечно-сосудистой системы и других органов, наблюдаться у профильных специалистов.
Для других наследственных нейропатий (группа НМСН, ННСПС) патогенетическая терапия не разработана, поэтому лечение ограничено симптоматической терапией, реабилитацией и рекомендациями по образу жизни:
- рекомендуется вести здоровый образ жизни, правильно и сбалансировано питаться, полностью отказаться от приема алкоголя, воздерживаться от вегетарианства;
- исключить, ограничить прием лекарственных препаратов с нейротоксическим действием;
- проводится коррекция ортопедических нарушений: подбор стелек, стоподержателей, коленоупорников, использование дополнительной опоры, кинезиотейпирование;
- регулярно, в поддерживающем режиме проводятся реабилитационные мероприятия: миостимуляция, массаж, растяжка, ЛФК.
.
Какой прогноз данного заболевания?
Прогноз определяется типом наследственной полинейропатии и многими сопутствующими факторами. Так, при НМСН 1 и 2 типа при позднем дебюте крайне замедленное развитие мышечной слабости приводит к тому, что больные приспосабливаются к своему дефекту и, несмотря на слабость и похудание кистей и стоп, могут выполнять ручную работу, свободно передвигаться. Даже при многолетнем течении сохраняется способность к самостоятельной ходьбе. Наследственная нейропатия со склонностью к параличам от сдавления также имеет относительно благоприятный прогноз при соблюдении рекомендаций по ограничению длительных статических поз и снижению потенциальных провокаторов компрессий нервов в повседневной жизни. НМСН III, V типов вызывают тяжелый двигательный дефект и в большинстве случаев приводит к выраженной инвалидности. При ранней диагностике наследственных нейропатий, ассоциированных с метаболическим дефектом (ТТР-САП, болезнь Рефсума, порфирия, болезнь Фабри) и назначении необходимой терапии возможно пролонгировать наступление инвалидизации и улучшить качество жизни.
В ФГБНУ НЦН проводят консультации сотрудники Центра заболеваний периферической нервной системы, сотрудники 5 неврологического отделения, профилем которого является диагностика и лечение наследственных заболеваний нервной системы. Консультации пациентов проводятся амбулаторно в рамках ОМС и на коммерческой основе.
На базе ФГБНУ НЦН проводится весь перечень необходимых для уточнения диагноза обследований, разработаны подходы к комплексной восстановительной терапии при наследственных полинейропатиях.
ЗАПИСЬ НА ПРИЕМ ПО МНОГОКАНАЛЬНЫМ ТЕЛЕФОНАМ
+7 (495) 374-77-76
+7 (495) 374-55-83
Дата издания: 23.04.2020